Folding@home (FAH or F@h) is a distributed computing project for simulating protein dynamics to understand human health/disease and contribute to the development of new therapeutics.
What’s the point?
Proteins are molecular machines that are responsible for most of the active processes that we associate with life. Some proteins are motors that power muscle contraction, others sense stimuli like light and sound, while others catalyze chemical reactions to break down food and build other molecules.
Like more familiar machines (e.g. cars), proteins have lots of moving parts that are essential to their ability to do useful things. We call the movements of these parts protein dynamics.
Many diseases, ranging from cancer to Alzheimer’s disease, result from aberrant protein dynamics. Unfortunately, it is often difficult, if not impossible, to watch these dynamics experimentally. That limits scientists’ ability to understand how protein dynamics are supposed to work, how they go wrong in disease, or how to fix protein dynamics.
Folding@home uses computer simulations to probe protein dynamics. These calculations are extremely computationally demanding, so we invite volunteers like you to use your spare computing resources to help us run more simulations.
What is folding?
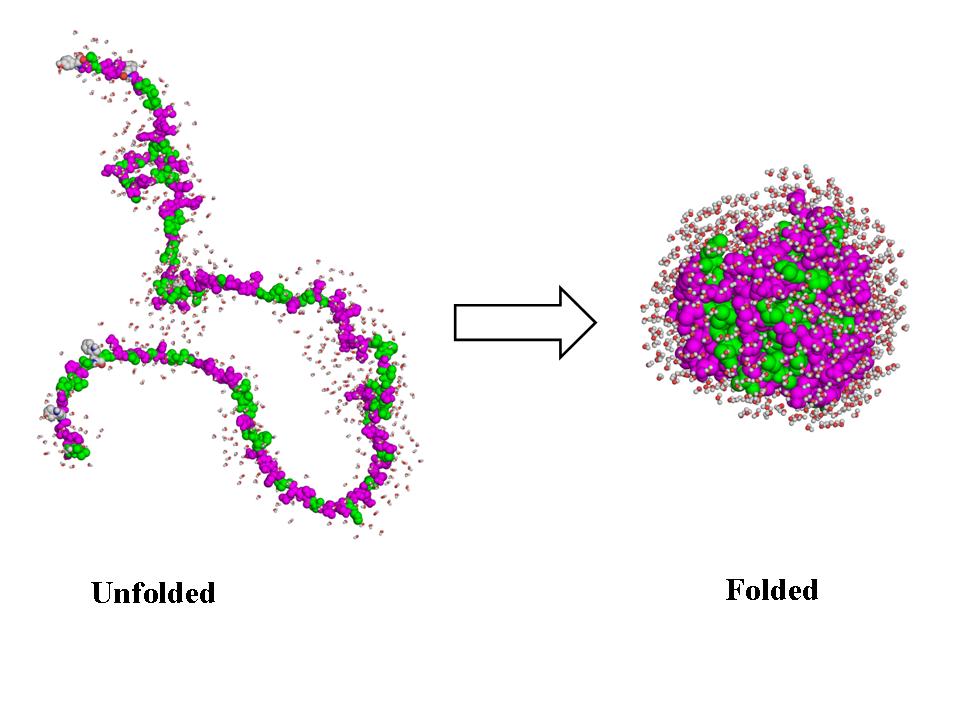
Folding can refer to the way proteins spontaneously build themselves. A protein consists of a linear chain of chemicals called amino acids, as in the extended structure on the left in the image below. The atoms in this chain push and pull on each other, ultimately causing the chain to fold up on itself into a compact structure that is capable of performing some function, like the structure on the right in the image below. The fact that many proteins can fold all on their own is amazing. Its like a pile of car parts putting itself together into a working car without a mechanic lifting a finger.
Folding can also refer to participating in Folding@home.
How does Folding@home work?
You can join our community of citizen scientists and help accelerate medical research by installing a piece of software called the client on your personal computers.
You control when and how much of your computing resources we use. For example, you can set the client up so that it only runs simulations when your computer is idle (e.g. when you’re sleeping). You can also specify which resources we can use. For example, if you have 8 CPU cores in your computer, you can tell the client to use 6 of them to run Folding@home, leaving you 2 cores for your uninterrupted use.
When the client starts, it reaches out to our servers and lets us know what resources your machine is ready to contribute. Our servers then send you a work unit (WU) that is well suited to your machine. A work unit consists of a protein structure (i.e. the coordinates of all the atoms in that protein) and instructions on how to simulate how these atoms move relative to one another in time. Essentially, your computer keeps asking the question “given how these atoms push and pull on each other, where will each of them be some small time in the future? over and over again. Your computer stores the positions of all the atoms at regular intervals. Once the work unit is done, you send the set of structures you generated (called a trajectory) back to our servers. Then your client requests a new work unit to complete. Work units are setup so that they take a few minutes to a few hours to complete, depending on how powerful your computer is.